Run of homozygosity (ROH) refers to contiguous regions of the genome where an individual is homozygous across all sites. This arises if the haplotypes transmitted from the mother and father are identical, having in turn been inherited from a common ancestor at some point in the past.The number and length of ROH reflect individual demographic history, while the homozygosity burden can be used to investigate the genetic architecture of complex disease. CEBALLOS F.C. et al. reviewed how ROH affects population history and trait architecture in Nature Review Genetics.
Origins of ROH and inbreeding depression
ROH arise when two copies of an ancestral haplotype are brought together in an individual: longer haplotypes inherited from recent common ancestors or shorter haplotypes from distant ones (background relatedness). Short ROH characterized by strong linkage disequilibrium (LD) among markers are not always considered autozygous but nevertheless are due to the mating of distantly related individuals. 
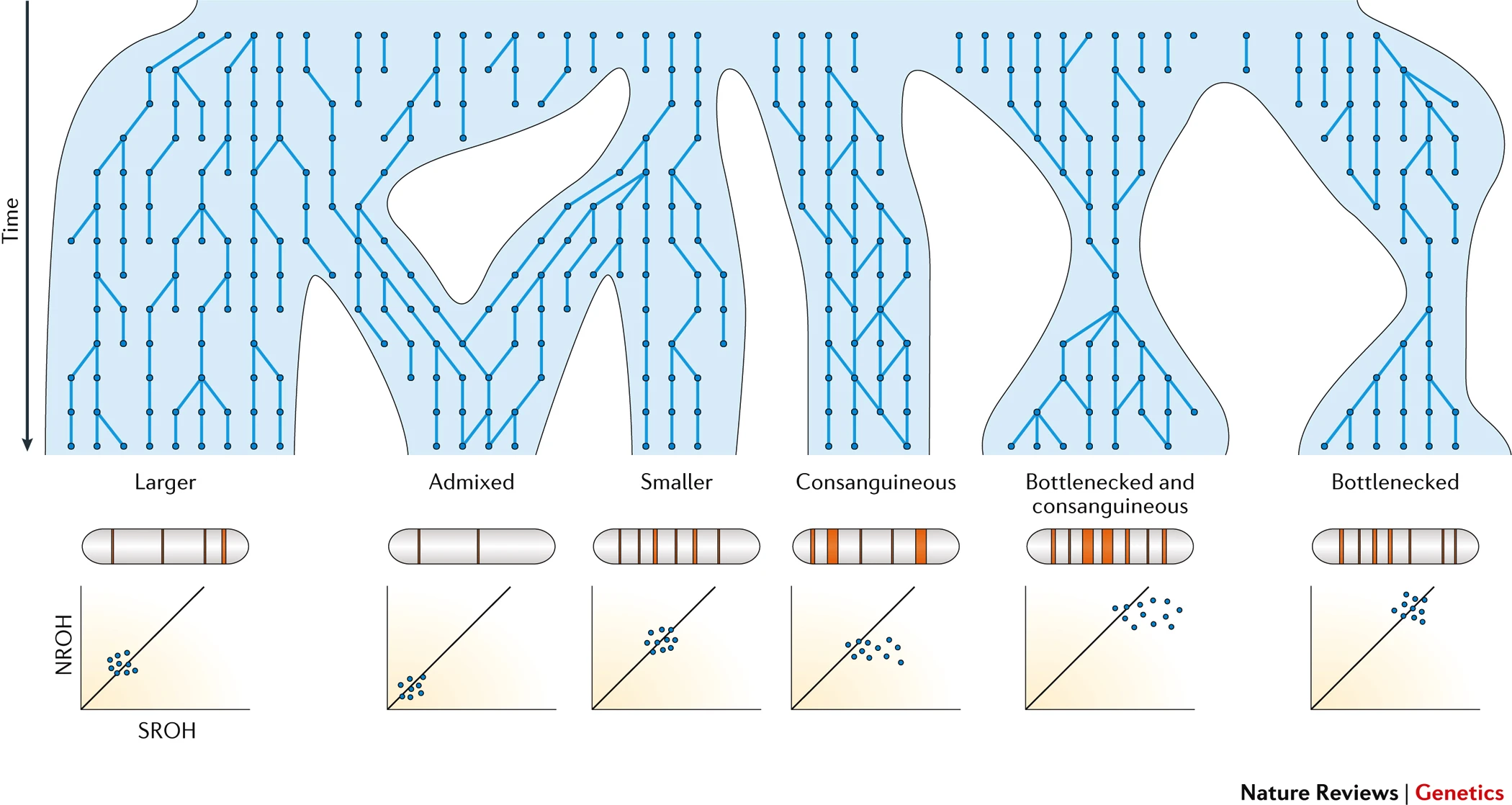
Different population histories give rise to divergent distributions of long and short ROH. The ROH complement of outbred populations is related to their effective population size, with smaller populations tending to have more ROH and larger populations fewer ROH. Admixed populations, on account of their more distant shared ancestry across two or more ancestral populations, have fewer ROH than their respective parental populations. Consanguineous communities, on the other hand, have much longer ROH than those seen in outbred populations owing to very recent pedigree inbreeding loops, whereas populations that have undergone a population bottleneck carry a greater number of shorter ROH than cosmopolitan populations, reflecting deeper parental relatedness. Finally, populations with both reduced effective population size in the past and recent inbreeding have the greatest burden of ROH.
The causal mechanism for inbreeding depression is only partly understood, but empirical evidence in a number of species suggests that it is due mostly to increased homozygosity for (partially) recessive detrimental mutations maintained at low frequency in populations by mutation–selection balance, although the contribution of some loci with heterozygote advantage (overdominance) maintained at intermediate frequencies by balancing selection cannot be disregarded. When dominant alleles at some loci decrease the trait value while others increase it, we do not expect any association with genome-wide homozygosity. However, if on average across all causal loci dominance is biased in one direction, for instance, to decrease the trait, we will see such an association. Such directional dominance arises owing to directional selection in evolutionary fitness-related traits. 
Empirical studies show that ROH are more enriched for homozygous deleterious variants than for non-deleterious variants. This emphasizes that ROH are important reservoirs of homozygous deleterious variation, although this is expected given the typically lower allele frequencies of deleterious variants compared with non-deleterious variants. Inbreeding increases the probability that a variant will be found in a homozygous state, so ROH are enriched for homozygotes at all allele frequencies. This enrichment is particularly strong for rare variants because a variant at frequency p is homozygous at frequency p2 outside ROH and at frequency p inside ROH (where p is the population frequency of the allele). Homozygotes thus occur (1/p) times more frequently inside ROH, so lower-frequency variants (including deleterious variants) are more strongly enriched. Theory also predicts that very strong inbreeding will in fact purge deleterious recessive alleles from the population as more copies are found in a homozygous state, and this has been observed in mountain and eastern lowland gorillas but not in human genome data.
Distribution of ROH
Correlation with pedigree inbreeding. The degree of individual inbreeding is measured using the inbreeding coefficient (F), the probability that an individual receives two alleles that are identical-by-descent at a given locus, which is also the expected proportion of the genome that is autozygous, for example, F = 0.0625 for the offspring of first cousins. The genomic inbreeding coefficient, FROH, measures the actual proportion of the autosomal genome that is autozygous — defined as the sum total length of ROH (SROH) over a specified minimum length threshold as a proportion of the total genome length. Another useful measure of ROH is the total number of ROH (NROH). FROH captures the total inbreeding coefficient of the individual, irrespective of pedigree accuracy or depth (or absence), within the resolution of the data set available (and hence the size of ROH that can be called).
***Distribution across the genome.***ROH are somewhat more common in regions of high LD and low recombination29 and are particularly prevalent on the X chromosome58 and regions of low genetic diversity. These observations are linked by low recombination: the X chromosome spends one-third of its time in the male germline, where (with the exception of the small pseudo-autosomal regions) it cannot recombine, and low-recombination regions have low SNP diversity. Recombination breaks up chromosomal segments over generations, and thus low-recombination regions allow greater persistence of long ancestral haplotypes and an increased chance that they come together to form ROH. Over and above this, there is a very uneven distribution along the genome, with a number of comparatively short regions with significant excesses of ROH — known as ROH islands — on each chromosome, as well as coldspots. These ROH islands dominate the population of ROH in typical outbred individuals, and while they are present in all populations, they are overshadowed by much longer ROH arising from recent pedigree loops in inbred individuals.
ROH and complex diseas
Quantitative traits. More consistency has been observed in studies of the association between SROH and quantitative traits, perhaps owing to the larger sample sizes, the use of common microarrays within study populations and the avoidance of unmatched controls. An exceptionally large study of up to 354,224 subjects found the regression coefficient between trait and the proportion of the genome in ROH to be −2.9 (0.2), −3.5 (0.7), −4.7 (0.6) and −4.6 (0.7) phenotypic standard deviations (standard errors in brackets) for height, forced expiratory lung volume in 1 second, cognitive ability and educational attainment, respectively.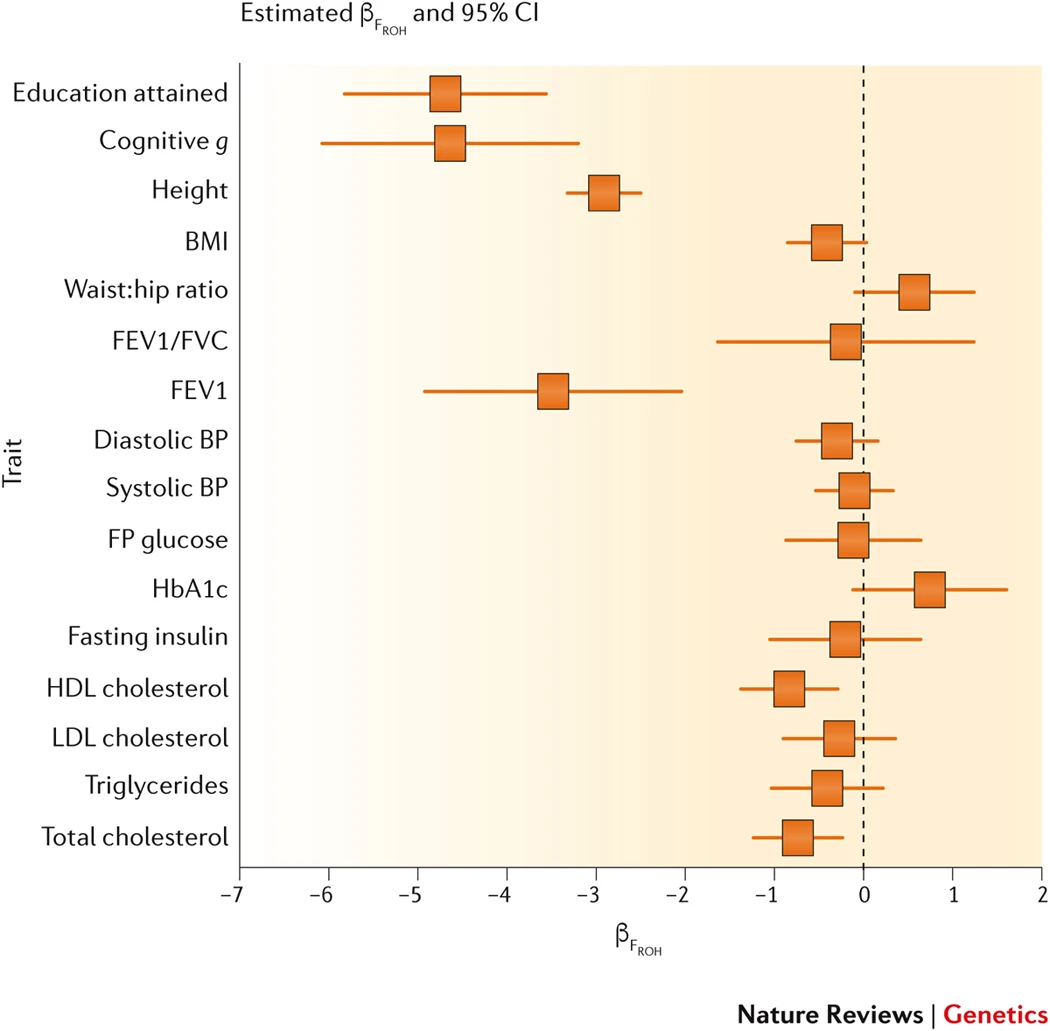
Individual ROH associations. The postulated model that homozygosity at rare, deleterious recessive alleles gives rise to directional dominance implies that there are specific loci within the genome giving rise to these effects. In principle, such loci should be discernible through a GWAS, although recessive models have been much less used than additive ones (and there is less power to detect rare variants than common ones). A slightly different approach identifies regions in which the occurrence of ROH is tested for association with the trait, aiming to detect a different class of variant from that found in GWAS. However, caution must be used in such exercises: multiple testing of large numbers of independent regions requires proper adjustment, and confounding by genome-wide homozygosity needs to be accounted for.